|
Table 1: Criteria measuring quality of experiment (report overall and as function of resolution)
|
|
Criterion | Target | Comments |
| Completeness | >95% of reflections to reported resolution | One of the most important factors in success. The low resolution or most intense reflections should not be missing selectively or overloaded. |
| Redundancy | >6-fold (including all wavelengths in MAD) | Important for rejecting outliers, which can hamper phasing and refinement. Lower redundancy must be accepted for crystals that suffer significant radiation damage. |
| Mean I/ɭ(I) for merged data | Higher values indicate better data | Good measure of signal-to-noise. |
| Rmeas1 | Lower values indicate better data | Measure of agreement between replicate observations, corrected for bias from number of replicates. |
|
Table 2: Criteria measuring quality of model (report overall and as function of resolution)
|
|
Criterion
|
Target for good structure
|
Comments
|
R=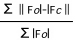
|
values slightly lower than Rfree are expected2
|
Traditional measure of data agreement, but subject to severe bias from over-fitting.
|
|
Rfree1
|
< 0.32 at 3.0ŕ resolution
< 0.26 at 2.5ŕ resolution
< 0.22 at 2.0ŕ resolution
|
Not subject to over-fitting bias, but subject to statistical error because of small number of observations used.
|
|
Residue density correlation3
|
No absolute target values
|
Regions significantly lower than mean are either incorrect or poorly ordered. Regions with out-of-register error often flanked by regions of low density correlation. |
|
Table 3: Criteria measuring the quality of experimental data
|
|
Criterion
|
Target (all tentative)
|
Comments
|
|
Completeness of backbone assignments
|
> 85%
|
Assignments to 1H�� 13C��, and 13C' are particularly valuable for analysis of secondary structure
|
|
Redundancy in assignment pathways
|
?
|
Should provide a useful measure of confidence in individual assignments that would give higher weight to structures determined from proteins labeled with 15N and 13C.
|
|
Completeness of NMR-observable proton contacts within 4 �
|
> 50%
|
(Doreleijers et al., 1999a)
|
|
Structural constraints per residue
|
> 12
|
For well-defined regions. These may be a mixture of various types of constraints
|
|
Table 4: Criteria measuring the quality of the model
|
|
Criterion
|
Target (tentative)
|
Comments
|
|
Atom coordinate rmsds within the set of conformers representing the structure
|
backbone atoms < 1ŕ
all atoms < 2ŕ
|
These are left deliberately loose so as not to encourage over-refinement of structures
|
|
NOE restraint violations (rmsd)
|
< 0.05ŕ
|
(Doreleijers et al., 1998)
|
|
Persistent NOE violations across the ensemble of structures
|
None > 0.5ŕ�
|
(Doreleijers et al., 1998)
|
|
Back calculation of NMR observables from the family of models
|
?
|
This could be NOESY spectra, chemical shifts, dipolar couplings, etc., as has been demonstrated in the literature. Consensus approaches are likely to develop in the next few years.
|
|
Table 6: NMR data items to be collected for a complete protein structure deposition
|
|
Authors
|
|
Citation
|
|
Molecular system studied
|
|
Natural source for the molecular system
|
|
Experimental source of molecular system
|
|
Sample contents
|
|
Sample conditions
|
|
NMR experiments carried out
|
|
Spectrometer used
|
|
Raw spectra
|
|
Spectral peak lists
|
|
Assigned spectral peak lists
|
|
Chemical shift referencing
|
|
Assigned chemical shifts
|
|
Coupling constants
|
|
Relaxation data (T1, T1rho, heteronuclear NOE, T2)
|
|
H-exchange rates (particularly if used to suggest H-bonding for structure
|
|
calculation)
|
|
<
Constraints listed by type
|
|
NOE
|
|
ROE
|
|
other distance constraints
|
|
H-bond
|
|
disulfide bond
|
|
residual dipolar coupling
|
|
torsion angle
|
|
J-coupling values
|
|
Chemical shifts (CA, CB, C', HA in particular)
|
|
Others (paramagnetic relaxation, salt bridges, ?)
|
|
Violated constraints (violations greater than 0.05 angstrom?)
|
|
Coordinates for a representative conformer
|
|
Coordinates for a family of conformers
|
|
Description of the software used to calculate conformers
|
|
Protocol used to calculate conformers
|